客户文章 | 奈瑟氏菌的分布式序列分型和聚类揭示了百年来流行性脑膜炎球菌的持续出现

1.简介
作者研发了新一代分布式cgMLST算法(distributed core genome multilocus sequence typing,dcgMLST)。新算法基于MD5哈希函数(数字“指纹”方法)获取基因组分型信息,不依赖中心数据库,实现去中心化的 cgMLST 分型。对149个苏州地区菌株(包括107个分离自40年前的菌株)进行测序,并整合全球69,994菌株建立了奈瑟氏菌dcgMLST技术,提出多个分类方案,可以极大提高奈瑟氏菌的物种鉴定、流行谱系鉴定和传播链鉴定效率。
奈瑟氏菌划分为39个种,其中包括10个潜在的新发现物种。
脑膜炎奈瑟氏菌被进一步分为168个地方谱系和3个大流行谱系。大流行谱系包括两个A血清群谱系和近期流行的W135谱系,三者在过去的百年中在全球造成至少9次大规模流行。
采用全新生信技术,对包含3个大流行谱系分别进行群体遗传学分析,首次系统重建了过去百年间流脑的全球传播模式,明确了我国流脑暴发和国际流行之间的密切关联。
提出全新的脑膜炎奈瑟氏菌流行模型,将其从地方谱系逐步演化形成大流行谱系并逐渐消亡的过程分为5个阶段,并总结各阶段特征。
采用dcgMLST对苏州的5次独立流脑暴发进行分析。我们发现传统流行病学分析极大高估了流脑病原体的传播次数,而基于高分辨率分析可以准确区分直接传播事件和密接人群本身的携带菌株。
2.引言
基于基因组学的全球病原体监控对传染病防治起关键作用,可在疾病暴发早期快速明确病原体来源和传播风险。但当前生物信息工具无力承担已有的大规模遗传数据的分析任务,亟待理论和技术创新。自 2013 年始核心基因组多位点序列分型技术(cgMLST)及配套的全球最大的病原体分型溯源数据库EnteroBase被建立。
cgMLST在各类病原体中选取数千个高度保守的持家基因,对其基因序列进行数字编码,是目前全球普遍采用的病原体分型技术。然而,现有cgMLST技术依赖单一中心数据库,存在计算瓶颈和数据安全风险。
为此作者团队研发了新一代分布式cgMLST算法(dcgMLST)。新算法基于MD5哈希函数获取基因组分型信息,不依赖中心数据库,实现去中心化的 cgMLST 分型,在满足疾控和科研要求的同时保障数据安全和高效计算。
奈瑟氏菌包括脑膜炎奈瑟氏菌和淋病奈瑟氏菌两种致病菌,每年导致约9000万次感染,特别脑膜炎奈瑟氏菌曾多次引起全球大流行。这些流行菌株的进化模式及其与地方性菌株的关系仍未得到充分探究。
借由新一代分布式cgMLST算法解析了奈瑟氏菌近百年的群体结构变化。进而实现了对近万余奈瑟氏菌基因组的大规模进化重建,成功解析流脑大流行谱系百年间的全球传播,并将其应用于流调中的短期传播链解析。这些全新进展将可以极大提高对流脑乃至其他细菌性病原体的认知和防控能力。
3.结果
3.1基于全球70000个奈瑟菌基因组数据集的分布式cgMLST方案和物种树
建立了69,994个奈瑟氏菌菌株的全球基因组数据集,包括来自GenBank的4411个组装基因组,来自NCBI SRA数据库的短reads组装的65,434个基因组,以及1975-2021年中国苏州脑膜炎奈球菌分离株的149个新基因组。最终收集的菌株来自英国(n = 14,536)、美国(n = 14,461)、挪威(n = 2511)、中国(n = 413)和其他90个国家。
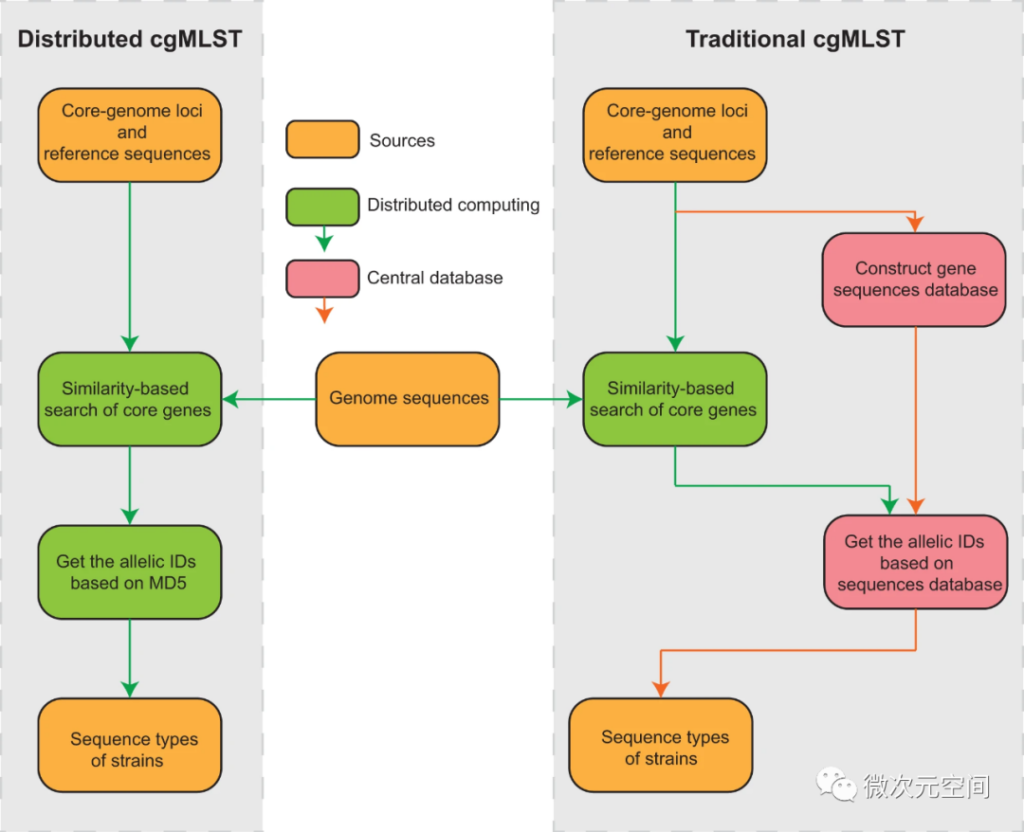
作者根据基因组间遗传距离挑选7630个具有代表性的基因组,并以此为基础建立了一个包含1149个核心基因位点的cgMLST方案,特别的,作者团队发明了一种算法,该算法将每个核心基因等位基因指定为基因序列的MD5哈希值,从而在没有中央数据库的情况下实现分布式cgMLST (dcgMLST)命名(图1)。此外,我们使用pHierCC将每个dcgMLST等位基因谱分配到一组1149个分层聚类水平,从HC0,即没有等位基因差异,到HC1148,其中除了一个等位基因外所有基因组都存在差异。
3.2基于dcgMLST聚类的物种重新划分
一直以来,奈瑟菌属下各个物种的分类被认为是错误和不完整的。为了修正物种名称,作者将代表性基因组细分为95%平均核苷酸一致性(ANI95%)的单链簇,这已被广泛用于许多细菌的细菌物种划分。然而,奈瑟菌的ANI95%聚类与种间不一致。例如,ANI95%不准确地将脑膜炎奈瑟菌和淋病奈瑟菌归为同一种。作者在物种树(图2)中发现了一个物种复合体的存在,并命名为NM复合体,它由许多人类特异性的脑膜炎奈瑟菌、淋病奈瑟菌、等多系群组成。作者确定了两个HC水平,HC1050和HC1130,分别形成类似于NM复合物内部和外部物种的簇。每个簇对应一个物种并且有极高的精度(ARI:0.99)。因此,HC1130和HC1050集群为传统分类和ANI提供了一种有效的替代方法,可以根据基因组序列将菌株快速分配到物种中。值得注意的是,作者还发现了6个HC1050集群和4个HC1130集群与任何现有物种都没有关联,这使得它们可能是新的物种,需要更详细的研究
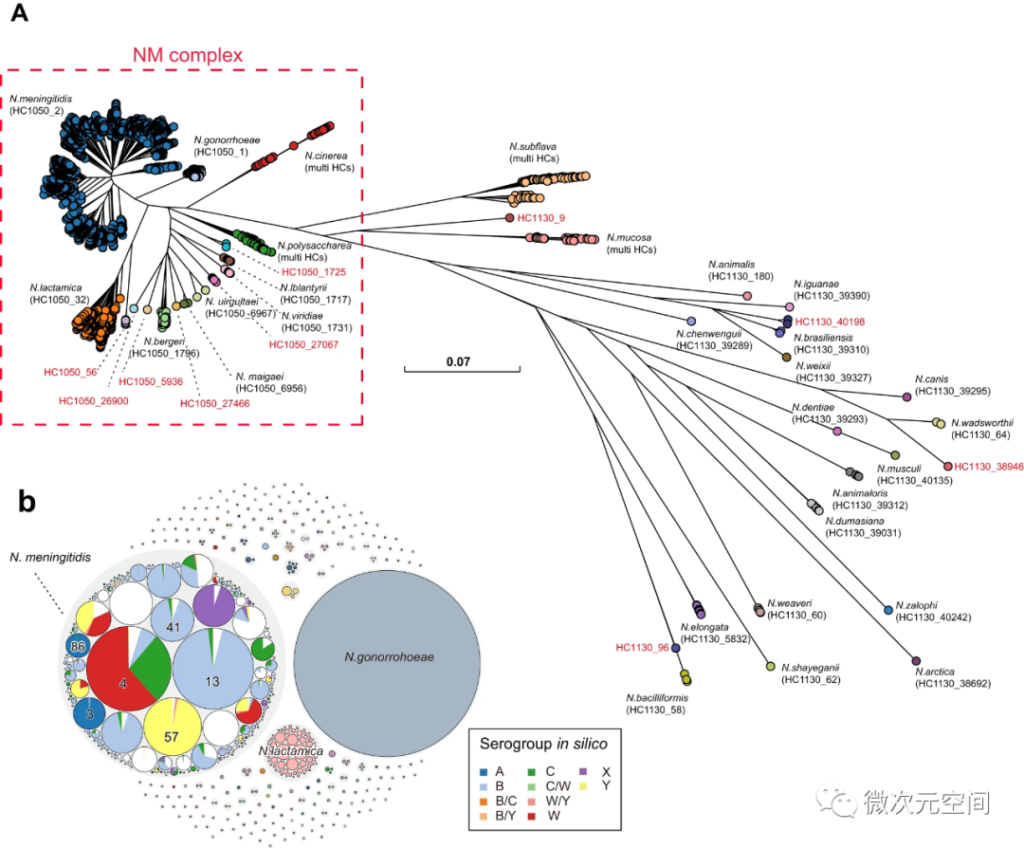
图2:奈瑟氏菌属群体结构及物种进化树
3.3 HC760揭示奈瑟菌种群的长期持久性
HC760聚类的Silhouette得分最高,并且与克隆复合体(Clonal Complex,CC)具有较好的一致性(ARI: 0.97)。整个奈瑟氏菌属中共包含570个HC760聚类簇。值得注意的是,所有淋病奈瑟菌都属于HC760_1,这表明它的遗传多样性比其他人类特异性奈瑟菌要低。此外,已知脑膜炎奈瑟菌血清群是高度可变的,并经历频繁的水平基因转移。唯一的例外是MenA,它只存在于两个HC760 (HC760_3和HC760_86)。在20世纪80年代之前,这两种HC760菌株占菌株总数的45%左右,并与上个世纪的大多数重大疾病暴发有关。在过去的40年中,这两种HC760的相对频率下降到仅3%,这可能与在流行地区应用MenA疫苗有关(图3a)。与此同时,2000年后欧洲MenB和MenY、非洲MenW和MenC、亚洲MenB、MenC和MenW的感染率分别增加了31-72%、<1%至64%、20-65%。
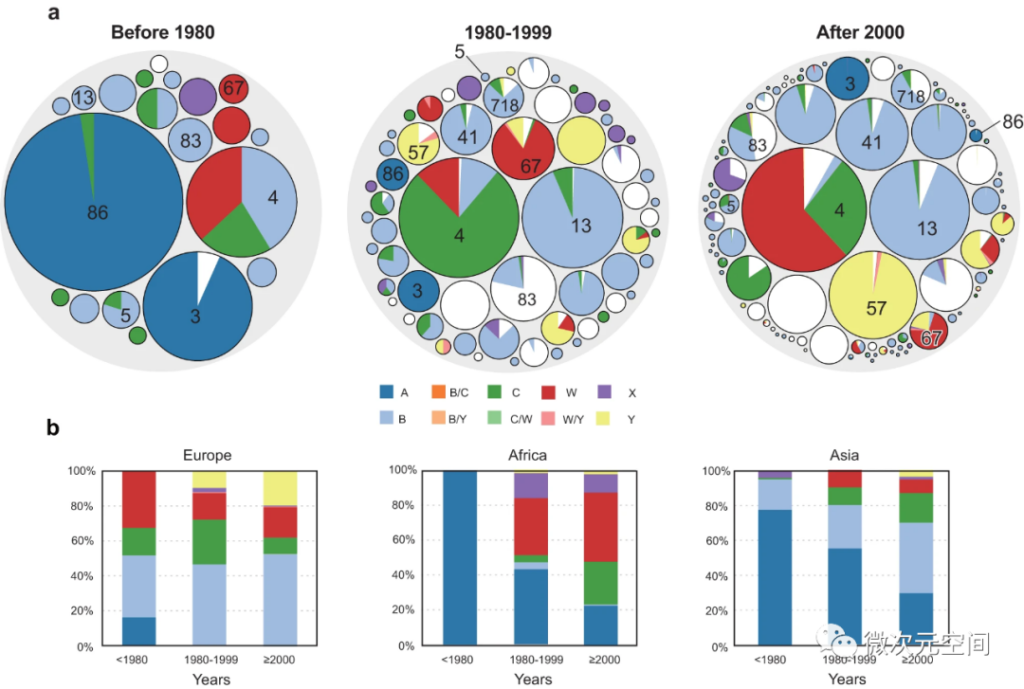
图3:脑膜炎奈瑟氏菌不同的血清群的种群变化
3.4 MenA HC760_3世系的进化轨迹
由于脑膜炎奈瑟菌血清群A (MenA)是上个世纪大多数重大暴发的原因,我们对该谱系进行了时间和地理系统发育分析,该谱系包含两个自然种群,其中一个起源于欧洲,并于20世纪50年代传播到北非,并于20世纪70年代至90年代传播到澳大利亚和南非。另一种起源于中国,在20世纪70年代至90年代期间至少三次传播到全世界。系统动力学推断分析表明,两个自然种群的有效种群大小有一定的变化,表明存在潜在的竞争关系。
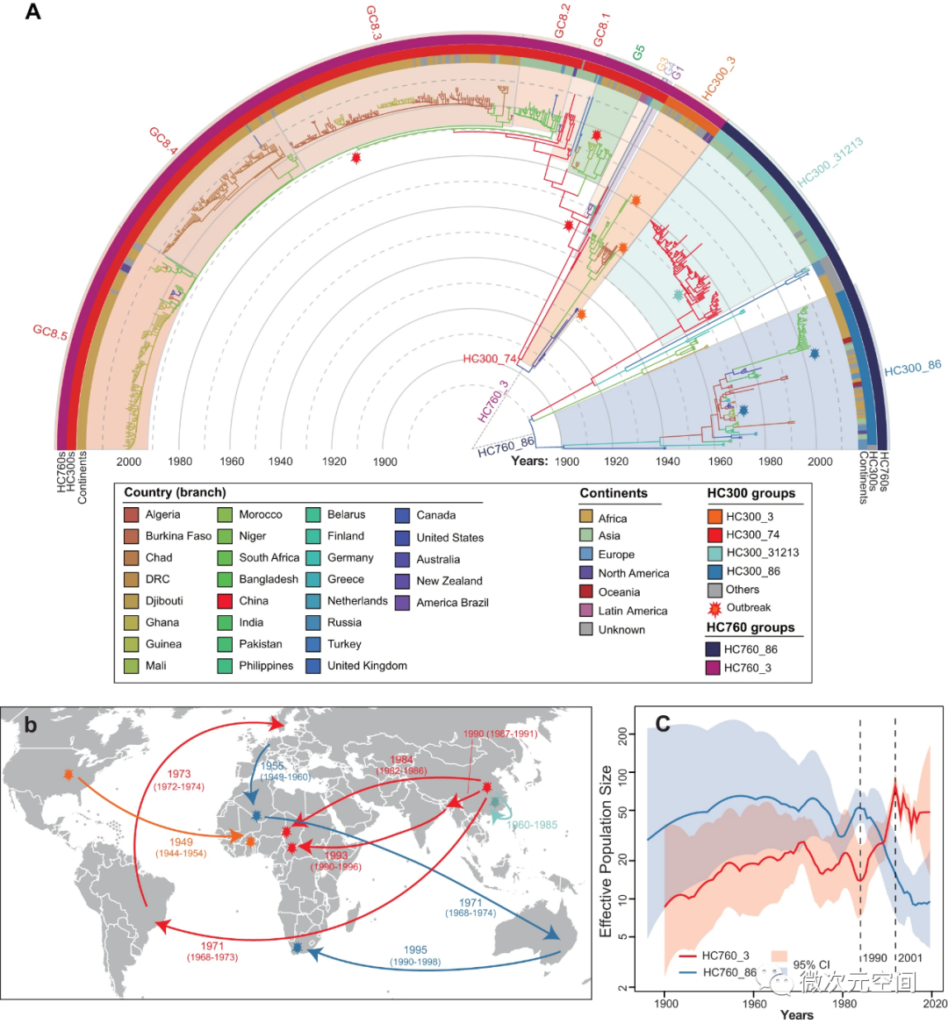
图4:MenA HC760_3世系的全球传播
3.5 新的全球流行病谱系:HC760_4的进化轨迹
在2000年著名的一次麦加朝觐造成暴发之后,W血清型的脑膜炎奈瑟氏菌成为今天主要的流行谱系。根据作者的分析,该谱系起源于欧洲,可分为C1、C2和W0-W3 等5个分支。与之前的报道一致,C-II的一个分支在1962年之前将其血清群转化为W,导致W-0分支的出现,该集群是其他MenW菌株的祖先。在1993年至1999年期间,分支W – 1多次传播到非洲,并于21世纪初在非洲流行。也是导致麦加朝觐爆发的病原体。

图5:HC760_4世系的全球传播
3.6 重建疾病暴发的传播链
为了证明dcgMLST可用于追踪疾病暴发,作者调查了5组有流行病学记录的苏州菌株。基因组数据与三例病例的流行病学记录部分一致,其中一些密切接触者携带的菌株与患者携带的菌株具有相同的HC10分类。另一些则显示了较大的遗传距离,表明非直接传播。这可能是因为人群中存在一定比例的无症状携带率,研究结果表明dcgMLST和分层聚类相结合可以识别传播链。
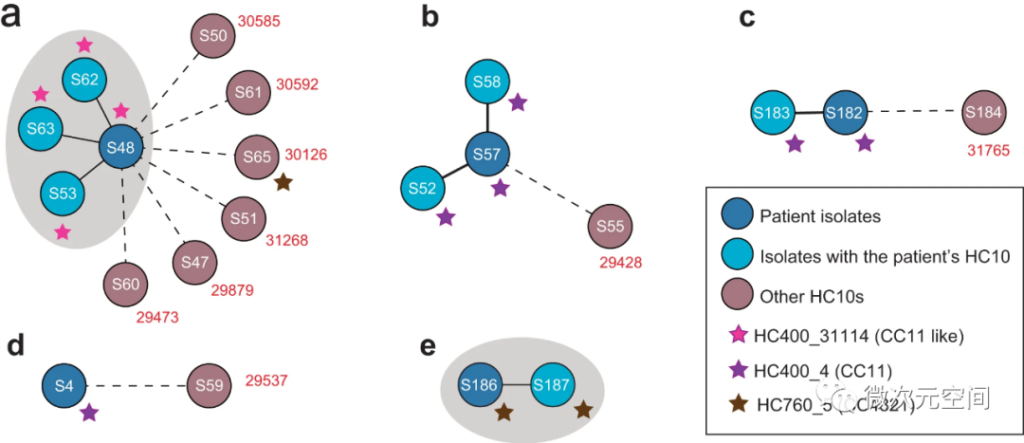
图6:利用HC10聚集性对5次疾病暴发的潜在传播进行评估
4.讨论
基于以上所有结果,我们提出了一个包含5个步骤的流行病学动态模型:首先,一些地方性流行的世系由于突变或重组而倾向于扩大其群体规模;接下来,这些新兴的谱系也分化成多个分支,其中一些传播到其他国家/地区。然而,由于与环境适应能力相关的一系列原因,这些早期传播中的大多数不会导致长期的疾病暴发。此外,一些进化支在其他地区定居下来,可能是由于一些超级传播事件的推动,导致了全球大流行病。最后,所有记录的脑膜炎奈瑟菌流行都逐渐被几十年来频率稳定的地方性谱系所取代
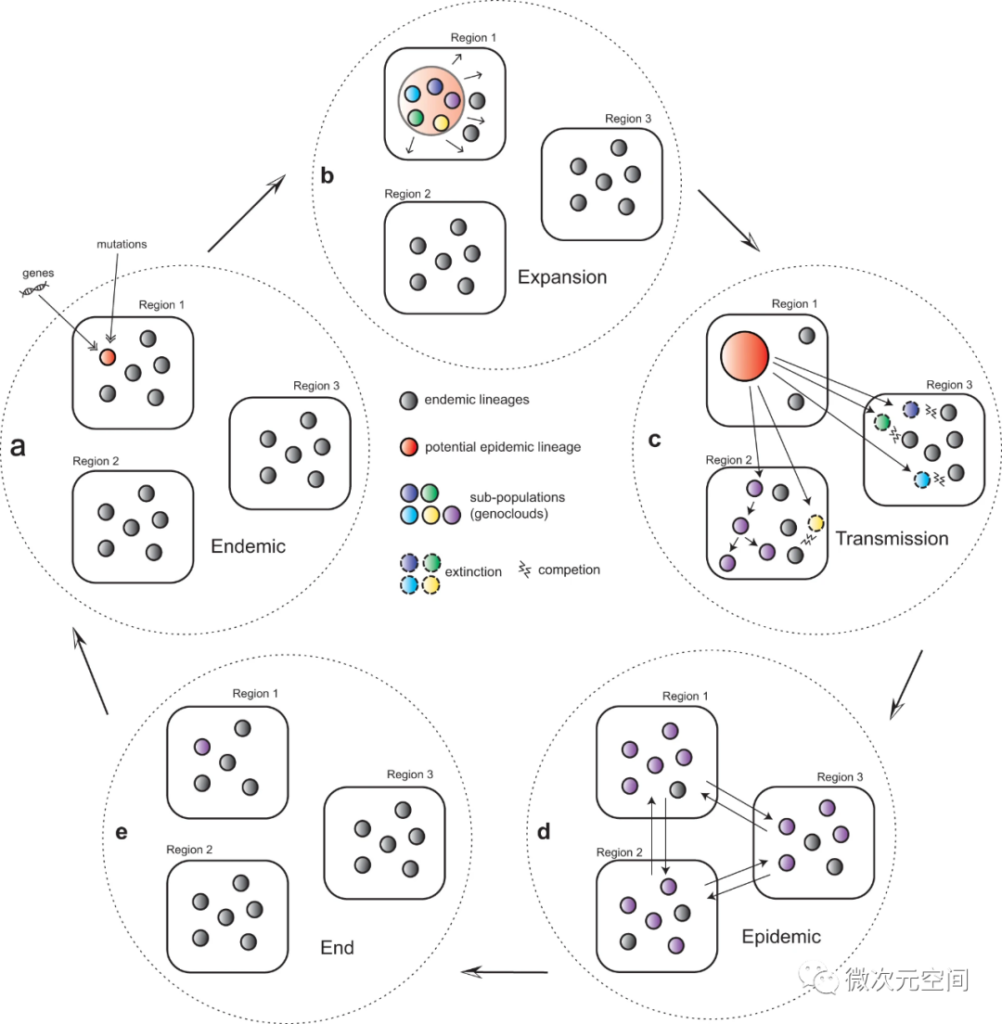
图7:新的流行病动力学模型
参考文献:
[1] Zhong, L., Zhang, M., Sun, L. et al. Distributed genotyping and clustering of Neisseria strains reveal continual emergence of epidemic meningococcus over a century. Nat Commun 14, 7706 (2023). https://doi.org/10.1038/s41467-023-43528-0